Dr. Md. Rashidul Haque
MBBS, FCPS (Psychiatry)
Assistant Professor
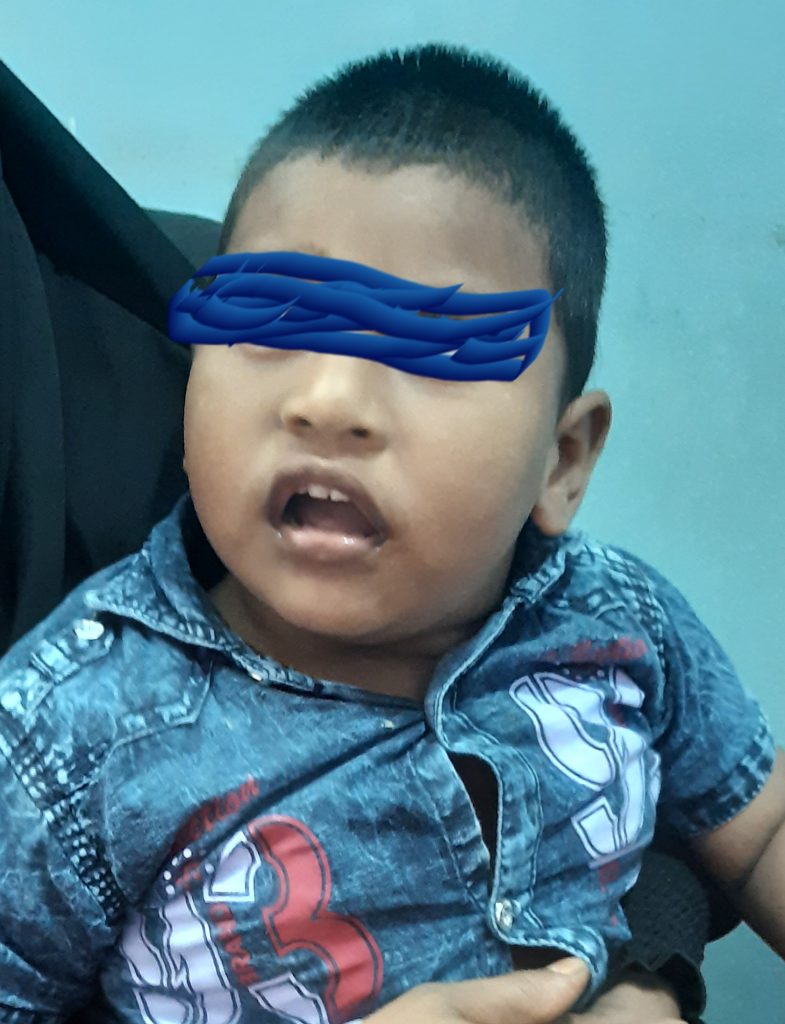
A 2 year 3 months old boy has attended in my chamber with a history of repetitive convulsions from the age of 1 year, inability to stand or sit or walk, can’t speak, sleep disturbance & low intelligence.
West syndrome is a constellation of symptoms characterized by epileptic/infantile spasms, abnormal brain wave patterns called hypsarrhythmia, and intellectual disability.
The spasms that occur may range from violent jackknife or “salaam” movements where the whole body bends in half, or they may be no more than a mild twitching of the shoulder or eye changes.
Epidemiology:
West syndrome is a rare neurological syndrome that can affect males and females. The X-linked form of West syndrome affects males more often than females. West syndrome has been estimated to affect 0.31 per 1000 live births in the United States. West syndrome accounts for approximately 30% of all cases of epilepsy affecting infants.
Signs & Symptoms
The average age of onset for epileptic spasms is at 6 months. Symptoms associated with West syndrome usually begin during the first year of life. Epileptic spasms are characterized by involuntary muscle spasms that occur due to episodes of uncontrolled electrical disturbances in the brain.
Each involuntary spasm typically begins suddenly and lasts for only a few seconds and occurs usually in clusters that can last over 10-20 minutes. Such episodes, which may occur upon awakening or after feeding. The seizures are characterized by sudden, involuntary contractions of the head, neck, and trunk and/or uncontrolled extension of the legs and/or arms. The duration, intensity, and muscle groups affected by seizures vary from infant to infant.
Most children will have regression of skills or delays in acquiring skills that require coordination of muscles and voluntary movements (psychomotor retardation).
The syndrome often develops into Lennox-Gastaut syndrome with mixed types of seizures that are difficult to control and is associated with intellectual disability.
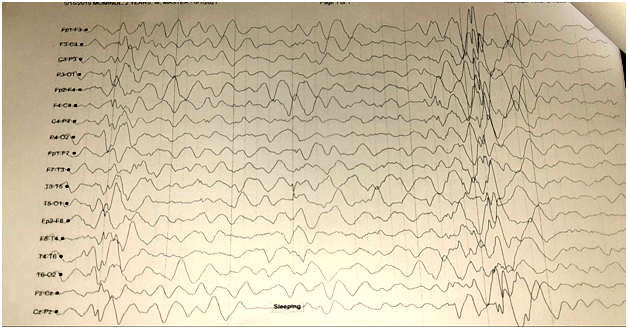
Fig2: EEG of Child with West Syndrome
Aetiology:
A specific cause for West syndrome can be identified in approximately 70-75% of those affected.
Any disorder that can lead to brain damage can be an underlying cause of West syndrome including trauma, brain malformations such as hemi-megalencephaly or cortical dysplasia, infections, chromosomal abnormalities such as Down syndrome, neurocutaneous disorders such as tuberous sclerosis complex (TSC), Sturge Weber syndrome, incontinentia pigmenti, different metabolic/genetic diseases such as pyridoxine deficiency, non-ketotic hyperglycemia, maple syrup urine disorder, phenylketonuria, mitochondrial encephalopathies and biotinidase deficiency, Otahara’s syndrome, and an abnormality (mutation) in the ARX gene or CDKL5 gene located on the X chromosome.
The most common disorder responsible for West syndrome is tuberous sclerosis complex. (TSC).
Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son.
Diagnosis:
Through history of child’s seizures and milestones of development (motor, speech & cognitive)
The first step is to characterize the patterns of brain activity through measurement with various devices.
Electroencephalography(EEG):
Infants with West syndrome also have a very abnormal electroencephalogram (EEG) with high amplitude, chaotic spike wave patterns (hypsarrhythmia).
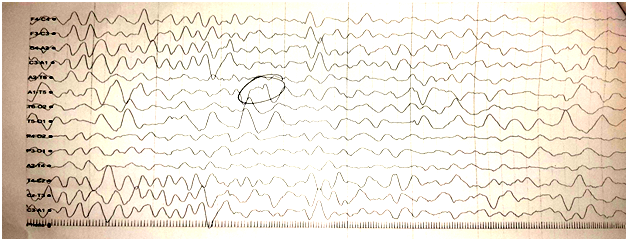
Fig3: EEG of the brain of a child of West Syndrome.
Computed Tomography (CT):
CT is also very good at showing areas of calcification that in some cases, may be essential for the diagnosis.
Magnetic Resonance Imaging (MRI):
The images can provide information concerning any malformations of the brain structures or other types of lesions commonly seen in epileptic spasms.
Molecular genetic testing:
It is available for mutations in the ARX and CDKL5 genes associated with X-linked West syndrome.
It is also available for the genes associated with tuberous sclerosis complex. Some genetic disorders will require cerebrospinal fluid (CSF) for genetic testing. Testing for nonketotic hyperglycemia may require a CSF sample to test for glycine and testing for mitochondrial diseases may require CSF to test for lactate.
A mutation in the STXBP1 gene has recently been noted in patients with Otahara’s syndrome as well. There are several genetic panels available that can test children of a certain age for a variety of genetic conditions that are seen in epilepsies such as epileptic spasms.
Infection as a cause of epileptic spasms may be determined by blood tests, urine tests and lumbar puncture.
A Wood’s lamp is used to examine skin for lesions with lack of pigment in order to determine if tuberous sclerosis is a possible diagnosis.
Differential diagnosis:
Symptoms of the following disorders can be similar to those of West syndrome.
- Epilepsy There are many different types of epilepsy and the exact cause is generally unknown. Epileptic spasm is a type of epilepsy.
- Lennox-Gastaut syndrome (LGS) is a rare type of epilepsy disorder that typically becomes apparent during infancy or early childhood. The disorder is characterized by seizures and, in many cases, abnormal delays in the acquisition of skills that require the coordination of mental and muscular activity (psychomotor delayes). Individuals with the disorder may experience several different types of seizures. Lennox-Gastaut syndrome may be due to, or occur in association with, a number of different underlying disorders or conditions.
- Myoclonic seizures can be seen in numerous types of epilepsies ranging from myoclonic epilepsy of infancy to Dravet syndrome or myoclonic astatic epilepsy and are often times confused with infantile spasms. These types of seizures are quick jerks of the arms and legs, faster than in infantile spasms and sometimes occur singly rather than in epileptic spasms that tend to occur in clusters.
- Myoclonus is a neurological movement disorder in which there are sudden involuntary muscle contractions. There are many different types of myoclonus including some that are hereditary. Other causes include lack of oxygen, viral, malignancies, and lesions of the central nervous system along with drugs and metabolic disorders.
Management:
Treatment may require the coordinated efforts of a team of specialists. Pediatricians, Child neurologists, Neuro-surgeons, Psychiatrist, other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
In some children, it is possible that treatment with anticonvulsant drugs may help reduce or control various types of seizure activity associated with West syndrome.
The most common medications used to treat epileptic spasms include adrenocorticotropic hormone (ACTH), prednisone, vigabatrin and pyridoxine. Vigabatrin was more effective in patients with tuberous sclerosis or cortical dysplasia compared to steroids.
More recently a multicenter European/Australian/New Zealand consortium (ISCC) found that hormonal therapy with vigabatrin is significantly more effective at stopping infantile spasms than hormonal therapy alone. The investigations in the US are ongoing for combination hormonal and vigabatrin therapy.
If these treatments are not successful, other medications such as benzodiazepines (for example, clobazam), valproic acid, topiramate, rufinamide and zonisamide may be used.
Ketogenic diet has also been successful at times in the treatment of epileptic spasms.
Finally, in cases where there is a malformation or tuberous sclerosis complex, epilepsy surgery may be helpful to control spasms.
Prognosis:
Approximately 1/3rd of children with West syndrome may develop recurrent epileptic seizures as they age.
Approximately another third of children with West syndrome will continue to have epileptic spasms at an older age.
The last third to quarter of patients will have spasms that resolve with time, usually in patients who have no clear etiology.